
Orthology
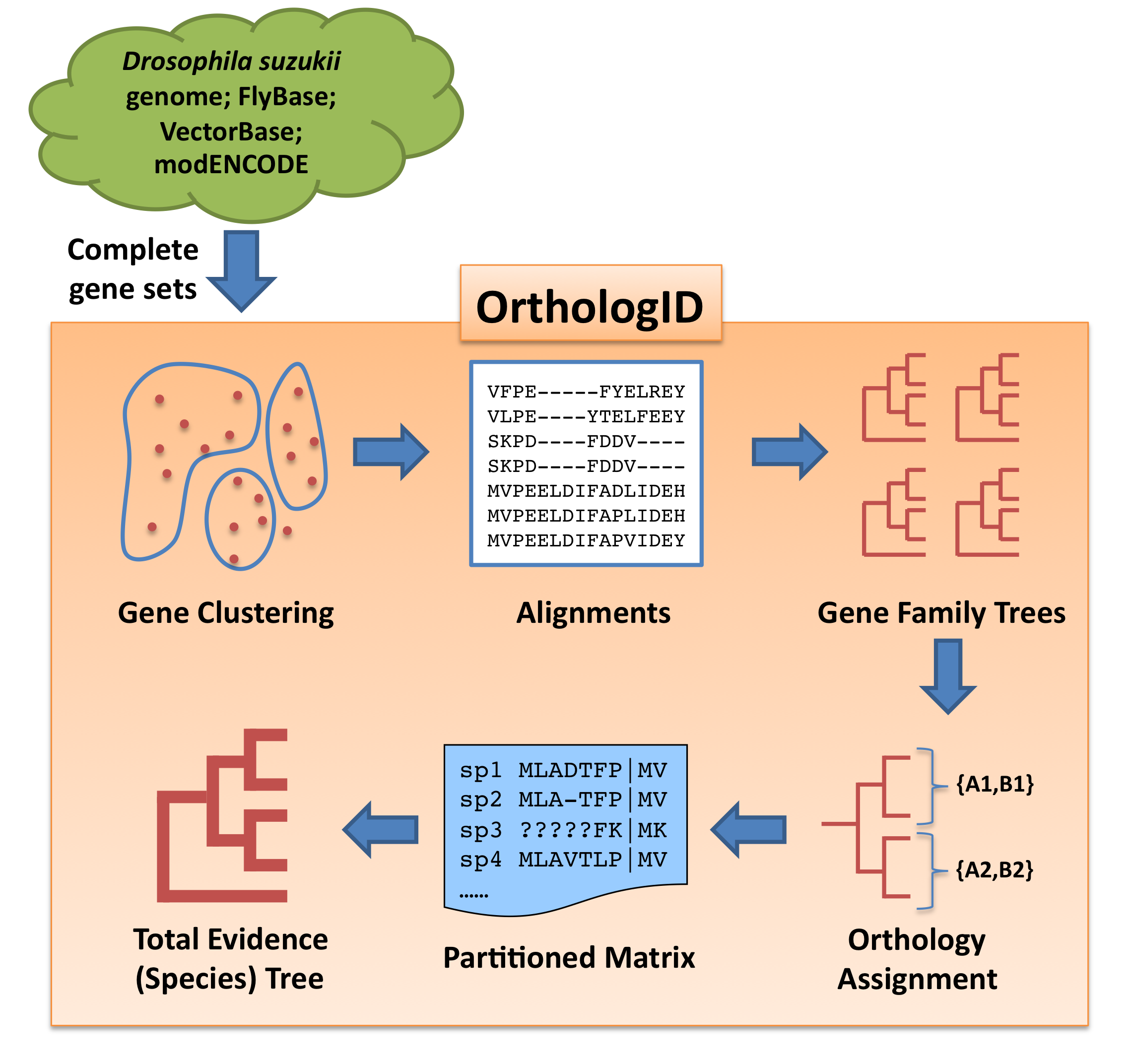 Gene orthology for all Drosophila species included in our analysis (D. suzukii, D. simulans, D. sechellia, D. yakuba, D. erecta, D. ananassae, D.pseudoobscura, D. persimilis, D. willistoni, D. mojavensis, D. virillis, D. grimshawi, D. melanogaster, D. takahashii, and D. biarmipes) and the outgroup Anopheles gambiae was evaluated using a pre-release version 2.0 of the OrthologID pipeline. Similar to the original version (Chiu et al. 2006, Bioinformatics 22(6): 699-707), OrthologID v2.0 takes complete gene sets from all ingroup and outgroup taxa and assigns them into gene clusters using the MCL algorithm. The complete gene set for A. gambiae and those for D. simulans, D. sechellia, D. yakuba, D. erecta, D. ananassae, D. pseudoobscura, D. persimilis, D. willistoni, D. mojavensis, D. virillis, D. grimshawi, and D. melanogaster were retrieved from VectorBase and FlyBase respectively and used as is. To generate the gene sets for D. takahashii and D. biarmipes, we downloaded the genome assemblies from GenBank and transcriptomes from the Drosophila modENCODE Project, and annotated their genomes using MAKER 2 – the same pipeline we used to annotate our assembly of the D. suzukii genome. OrthologID then performed sequence alignment for each gene cluster using MAFFT, generated a parsimony gene tree for each gene cluster, and extracted one or more sets of orthologous genes according to the gene tree topology. Finally, OrthologID assembled the ortholog sets into a partitioned matrix for phylogenetic analysis. Using gene sets from the 16 species as input, OrthologID recovered 13,941 sets of orthologs with at least 4 ingroup species from 13,264 gene clusters. Among the identified ortholog sets, 5,322 of them have representation in all ingroup taxa.
Gene orthology for all Drosophila species included in our analysis (D. suzukii, D. simulans, D. sechellia, D. yakuba, D. erecta, D. ananassae, D.pseudoobscura, D. persimilis, D. willistoni, D. mojavensis, D. virillis, D. grimshawi, D. melanogaster, D. takahashii, and D. biarmipes) and the outgroup Anopheles gambiae was evaluated using a pre-release version 2.0 of the OrthologID pipeline. Similar to the original version (Chiu et al. 2006, Bioinformatics 22(6): 699-707), OrthologID v2.0 takes complete gene sets from all ingroup and outgroup taxa and assigns them into gene clusters using the MCL algorithm. The complete gene set for A. gambiae and those for D. simulans, D. sechellia, D. yakuba, D. erecta, D. ananassae, D. pseudoobscura, D. persimilis, D. willistoni, D. mojavensis, D. virillis, D. grimshawi, and D. melanogaster were retrieved from VectorBase and FlyBase respectively and used as is. To generate the gene sets for D. takahashii and D. biarmipes, we downloaded the genome assemblies from GenBank and transcriptomes from the Drosophila modENCODE Project, and annotated their genomes using MAKER 2 – the same pipeline we used to annotate our assembly of the D. suzukii genome. OrthologID then performed sequence alignment for each gene cluster using MAFFT, generated a parsimony gene tree for each gene cluster, and extracted one or more sets of orthologous genes according to the gene tree topology. Finally, OrthologID assembled the ortholog sets into a partitioned matrix for phylogenetic analysis. Using gene sets from the 16 species as input, OrthologID recovered 13,941 sets of orthologs with at least 4 ingroup species from 13,264 gene clusters. Among the identified ortholog sets, 5,322 of them have representation in all ingroup taxa.
As part of the output of the OrthologID automated comparative genome analysis pipeline, we have made available the sequence alignment and parsimony tree for all gene clusters in SpottedWingFlyBase‘s Gene Family reports. The gene trees can be downloaded as Newick tree files, which can be visualized using any tree viewing software, e.g. FigTree. The alignments can be downloaded as zipped FASTA files and visualized using most alignment and sequence analysis packages, e.g. Jalview.